biOpen 2de AbOrygen
biOpen 2 es una innovadora e intuitiva aplicación de biología molecular diseñada para simplificar y mejorar las labores de investigación. Entre sus características detacan la integración dinámica y directa de herramientas de análisis de secuencias, sus potentes y exclusivas funciones de visualización, y su intuitiva interfaz de gestión de proyectos.
|
biOpen® is a new and unique sequence analysis and structure visualization software for Mac OS X based on dynamic integration of analysis tools.
Features:
- Extensive modularity
biOpen® gives you the freedom to pick the sequence analysis tools that meet your needs and budget. All selected tools work in a fully integrated environment with powerful display and project management capabilities.
- Easy evolution of your configuration
You can get new sequence analysis tools and integrate them in your biOpen® configuration when needed.
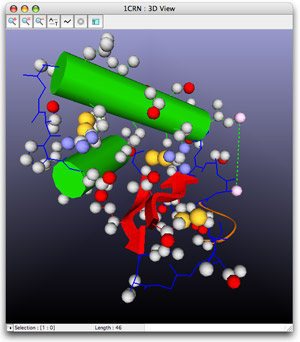
- 3D visualization
The relations between the sequence and its structure can be studied with one single software: simply select a part of the sequence and see it light up on the structure.
- Powerful display features
Fully synchronized views, dynamic display of sequence analysis results, automated extraction and display of annotations from GenBank sequence files, and even more.
- Project management
Organize and save your sequences, structures and results very easily. Import most popular file formats, including DNA Strider. Projects can be exchanged with your work colleagues.
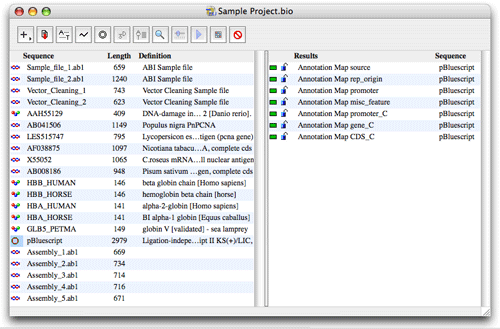
In biOpen®, sequences are organized in projects. From the project window, you can import, open, export and organize sequences, structures and analysis results very easily. Projects including all your data can be exchanged with your work colleagues.
The left part of the Project window shows the sequences in the project. Sequence type, name, size (base pairs or amino acids), definition, accession number and organism are displayed.
The right part of the Project window shows the results from sequence analysis in the project. Results available for a sequence are displayed by clicking on the sequence. If several sequences are selected, all results available for the selected sequences are displayed.
- Graphical export
biOpen® views can be simply exported in your favorite word processor or presentation software. You just have to perform a selection and drag and drop it to your document. It can then form an integral part of your project report or presentation.
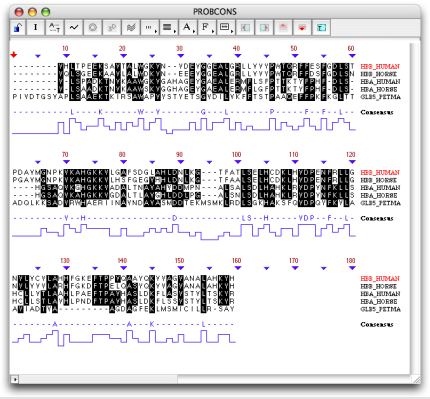
- Multiple sequence alignment editor
The multiple sequence alignment window can be opened by double-clicking on the multiple sequence alignment result in the project window or by selecting the result from the sequence editor or graphical window. The multiple sequence alignment view shows the alignment of the sequences, with the consensus sequence and graph displayed at the bottom.
The Use black shading option allows you to color background in black and residues in white.
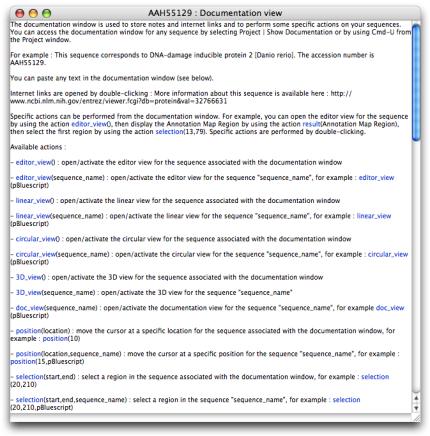
- Documentation
The documentation window is used to store notes and internet links and to perform some specific actions on your sequences. You can paste any text in the documentation window. Internet links are opened by double-clicking.
Specific actions can be performed from the documentation window. For example, you can open the editor view for the sequence by using the action editor_view(), then display a result by using the action result(result_name), then select a region by using the action selection(start,stop). Specific actions are performed by double-clicking. You can access the documentation window for any sequence by selecting Project | Show Documentation or by using Cmd-U from the Project window.
- Accessing Internet Databases
The Internet Tools menu offers links to useful databases and analysis tools. Selecting a page from the menu will open it with your default Internet browser.
When required, sequences can be dragged and dropped from biOpen to the appropriate forms in the Internet pages. To download a sequence or structure file, hyperlinks can be dragged and dropped from the browser to the project window.
- Creating a scientific report
All sequence views (editor, graphical or 3D) can be simply exported. You just have to perform a selection and to drag and drop it to your document. It can then form an integral part of your project report or presentation.
Tools for biOpen
- Contig Assembly
Contig Assembly enables you to assemble fragment data from sequencing projects and to create complete assemblies, called contigs. Each contig is a multiple alignment of contiguous, overlapping sequences. Contig Assembly also allows you to align one or more sequencing files to a reference sequence.
- Open Reading Frame
Open Reading Frame (ORF) analysis enables you to find and display open reading frames bordered by potential start and stop codons.
- Restriction Enzyme
Restriction Enzyme analysis is used to locate restriction enzyme cut sites in a nucleic acid sequence and predict the fragments that would result from single or multiple digests.
- PCR
The PCR tool analyzes a template DNA sequence and chooses primer pairs for the polymerase chain reaction (PCR). Two methods are available: the "Product size" method allows to search for primers according to a specified product size range, and the "2 flanking regions" method allows to search for primers surrounding the sequence region to be amplified.
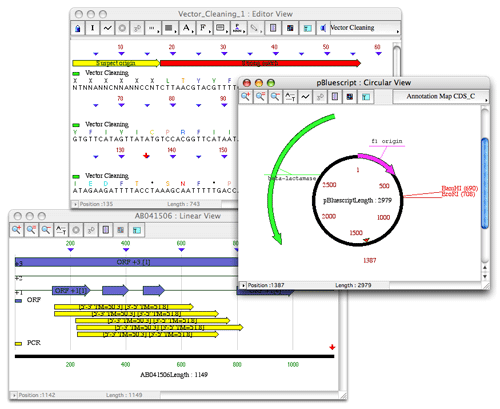
- Vector Cleaning
Vector Cleaning uses the BLAST algorithm for detection and removal of vector contamination in a nucleic acid sequence. Vector cleaning groups matches to vector sequences into three categories: Strong match, Moderate match and Weak match. Matches are categorized according to the expected frequency of an alignment with the same score occurring between random sequences. Any segment of fewer than 50 bases between two vector matches or between a match and an end is considered as a segment of Suspect origin.
- Physico-chemical profiles
In this module you will find tools allowing to calculate and plot several physico-chemical profiles. Tools available are accessibility, antigenicity, flexibility, hydrophilicity and hydrophobicity.
- RBD
RBD is a fast method for predicting residues involved in protein interaction sites. The function identifies linear stretches of sequences as "Receptor-Binding Domains" (RBDs) by analysing hydrophobicity distribution.
- TMpred
TMpred makes a prediction of membrane-spanning regions and their orientation. It is based on the statistical analysis of TMbase, a database of naturally occuring transmembrane proteins and their helical membrane-spanning domains (Hoffman and Stoffel, 1993).
The prediction is made using a combination of several weight-matrices for scoring. TMpred will predict possible inside-to-outside and outside-to-inside helices and will suggest models (prefered and alternate) for transmembrane topology. These suggestions are purely speculative and should be used with extreme caution since they are based on the assumption that all transmembrane helices have been found.
- Pairwise sequence alignment using dynamic programming
Global pairwise NW uses the algorithm of Needleman and Wunsch (1970) to find the optimum alignment of two complete sequences that maximizes the number of matches and minimizes the number of gaps. Global pairwise NW is used for aligning two sequences over their entire length.
Local pairwise SW uses the algorithm of Smith and Waterman (1981) to make an optimal alignment of the best segment of similarity between two sequences. Local pairwise SW is used for aligning the best matching subsequences of two sequences.
- Pairwise sequence alignment using BLAST and MEGABLAST
Blast2seq uses the BLAST algorithm for pairwise DNA-DNA or protein-protein sequence comparison. Blast2seq finds multiple local alignments between two sequences, allowing the user to detect homologous protein domains or internal sequence duplications.
Megablast2seq uses a greedy algorithm for the nucleotide sequence alignment search. This program is optimized for aligning sequences that differ slightly as a result of sequencing or other similar errors. When larger word size is used, it is up to 10 times faster than more common sequence similarity programs. Megablast2seq is also able to efficiently handle much longer DNA sequences than Blast2seq.
- Multiple sequence alignment using MUSCLE
MUSCLE was developed by Dr Edgar (2004) for creating multiple alignments of nucleic acid and protein sequences. Elements of the algorithm include fast distance estimation using kmer counting, progressive alignment using log-expectation score, and refinement using tree-dependent restricted partitioning. MUSCLE provides significant improvements in both speed and accuracy compared to CLUSTALW and T-Coffee.
- Multiple alignment of protein sequences using PROBCONS
PROBCONS was developed by Dr Do et al. (2005) for creating multiple alignments of protein sequences. Using a combination of probabilistic modeling and consistency-based alignment techniques, PROBCONS has achieved the highest accuracies of all alignment methods to date. On the BAliBASE benchmark alignment database, alignments produced by PROBCONS show statistically significant improvement over current programs, containing an average of 7% more correctly aligned columns than those of T-Coffee, 11% more correctly aligned columns than those of CLUSTAL W, and 14% more correctly aligned columns than those of DIALIGN.
|
|
|
|